
Genetically Modified Mice
Welcome to Ozgene
Genetically modified mice for research

OUR SERVICES
Our proven track record with genetically modified mice
spans more than two decades






Humanized mice up to 240 kb
Reduced project timelines and costs with OzBIG
Ozgene offers a service called OzBIG for robust creation of large, gene-targeted replacement or safe harbor targeted models. This system enables routine generation of humanized mice with very large (20 to 240 kb) genomic replacements or insertions. Ozgene are pleased to announce that they are working in partnership with Gen H in supporting our OzBIG capabilities. Read more here!
What is goGermline™?
goGermline is a fast and efficient way to generate genetically modified knockout, knock-in and targeted transgenic mice. goGermline eliminates competition with ‘host’ embryos, resulting in 100% embryonic stem (ES) cell-derived offspring with significant benefits to project timelines and animal welfare. To experience the goGermline benefits first-hand, enquire about a mouse project with us or order goGermline embryos to trial them in your facility.
How long does it take to generate mouse models?
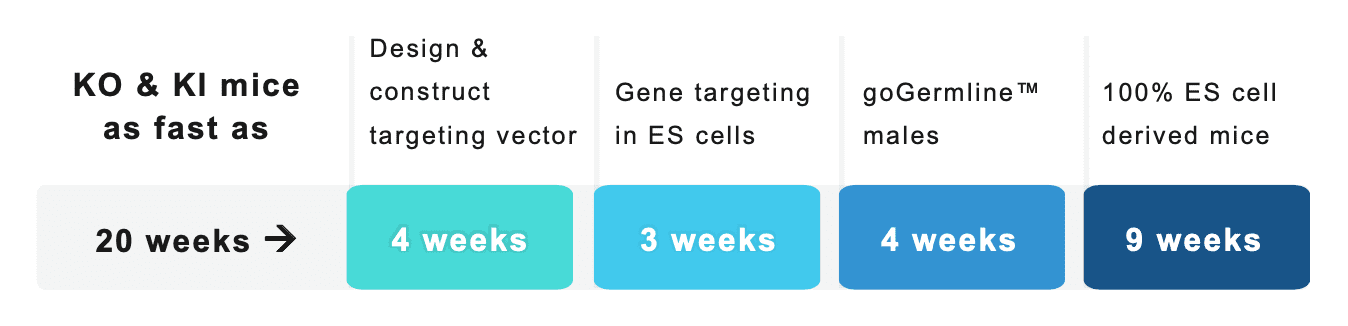
“No project queues – get started today!”
NEWS & BLOG
What’s making news?
Stay up to date with the latest Ozgene news, research features, scientific investigations, blog and podcast, all in one place.

Visits, preorders & feedback
The 31st of May will mark one year since Ozgene ARC assumed the operations of the former Animal …

Hot start to 2024
Thank you to all of our customers for your understanding and flexibility given the recent heat waves here …
Get in touch
We offer personalised services for your research needs. Request a free quote today.
Please fill out the form and we will respond to your query within two business days. Alternatively, visit our contact page for more ways that you can get in touch with us.